


根据斯坦福大学人本人工智能研究所(HAI)发布的《2025年AI指数报告》1,人工智能(AI)在医疗器械领域的应用取得了显著进展,尤其在医学影像、临床试验、辅助诊断等方面展现了强大的潜力。2023年,FDA批准的AI医疗器械数量从2015年的6款激增至223款,反映了AI在医疗保健领域的广泛应用。AI驱动的技术不仅在癌症检测和高风险患者识别方面超越了医生,还在蛋白质序列优化、合成数据生成和疾病预测等领域表现出极大潜力。
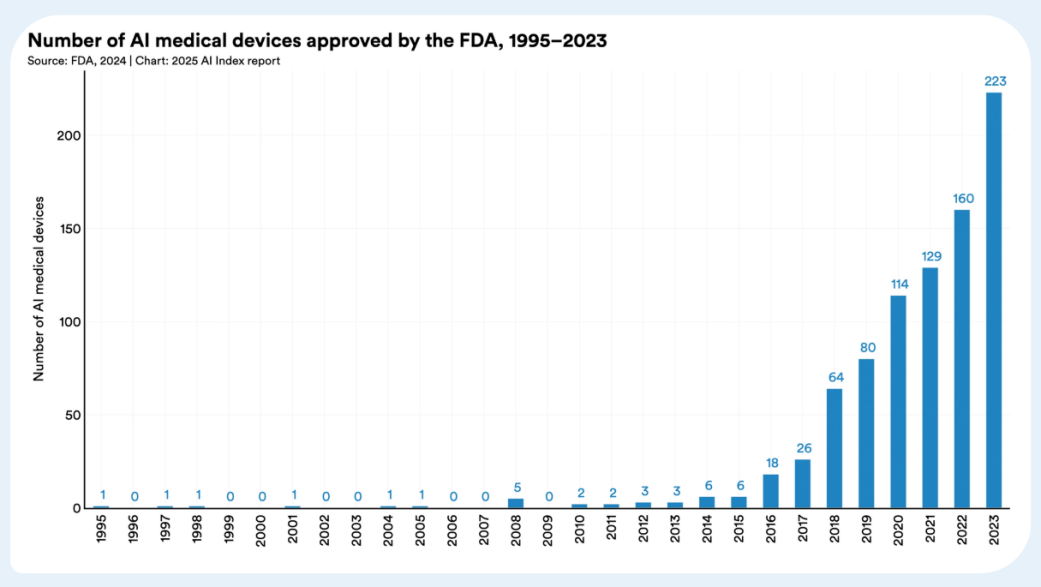
对于正将人工智能/机器学习(AI/ML)技术融入医疗器械产品的中国药械企业而言,在全球化布局过程中正面临多重合规挑战。产品市场准入许可、跨境数据流动监管等关键环节,构成了企业出海必须突破的合规矩阵。为系统解析全球主要市场的监管框架,本系列文章将深度解构三大核心市场的监管范式:上篇聚焦美国FDA基于SaMD框架的AI医疗软件认证体系,中篇剖析欧盟MDR法规下AI医疗器械的临床评估要求,下篇则立足中国《人工智能医用软件产品分类界定指导原则》。通过比较法视角的监管要义解读,助力企业建立符合国际标准的AI医疗器械产品合规体系。
一、FDA传统监管途径
对于人工智能医疗器械,美国食品药品监督管理局(Food and Drug Administration, “FDA”)的监管一方面在近年来对人工智能/机器学习(Artificial Intelligence and Machine Learning, “AI/ML”)技术创建了一系列附加考虑要素,另一方面其监管逻辑仍延续传统的医疗器械分类体系和审批路径,且后者是解析人工智能医疗器械产品合规的底层基础。
(一)基于风险等级的分类
FDA对医疗器械的传统监管思路遵循风险分层原则,其法律依据源于《联邦食品、药品和化妆品法案》(Federal Food, Drug and Cosmetic Act, “FD&C Act”)第513条确立的器械分类制度。根据该制度,器械的监管强度与其风险等级呈正向比例关系,即对患者构成最高风险的器械必须提出最严格的要求,以证明其安全性和有效性,然后才能在美国合法销售。具体表现为三类递进式监管框架(见下表):
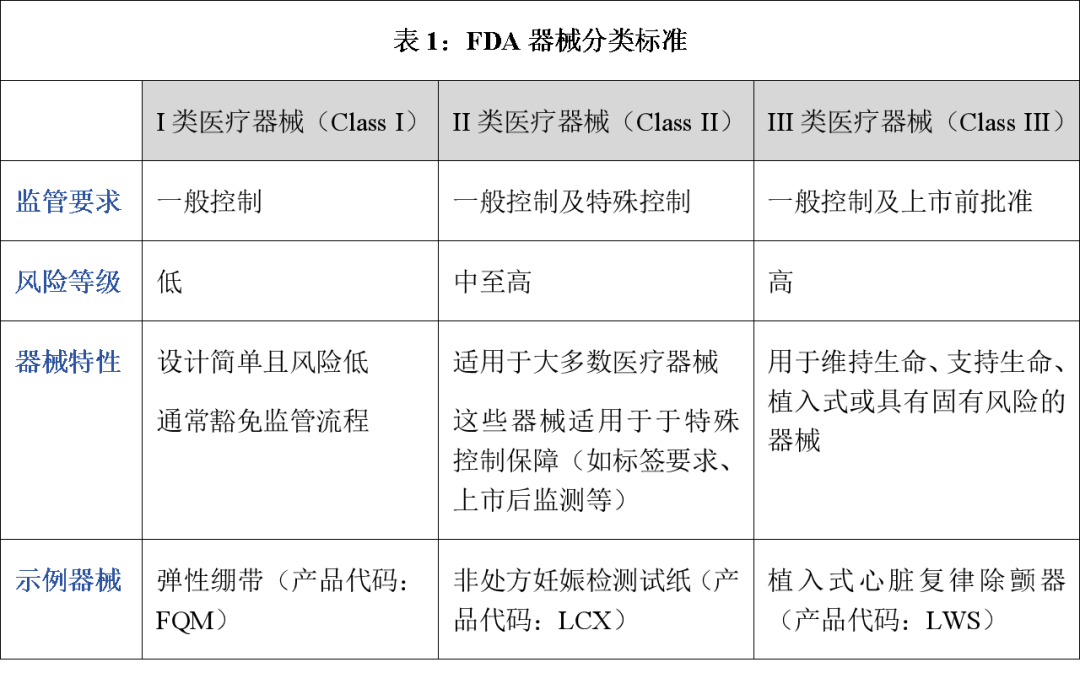
(二)具体监管审批路径
针对上述三种不同风险等级的医疗器械产品,FDA设置了差异化的监管审批路径:包括针对罕见病治疗器械的人道主义豁免(Humanitarian Device Exemption, “HDE”)程序、基于实质等效原则的上市前通知(Pre-market Notification, “PMN/501(k)”)简化路径,以及高风险器械的上市前批准(Pre-market Approval, “PMA”)强制要求,同时揭示了通过513(g)重分类申请与新型医疗器械分类请求(De Novo Classification Request, “De Novo”)机制实现监管要求动态适配的特殊通道(具体流程参见下图)。
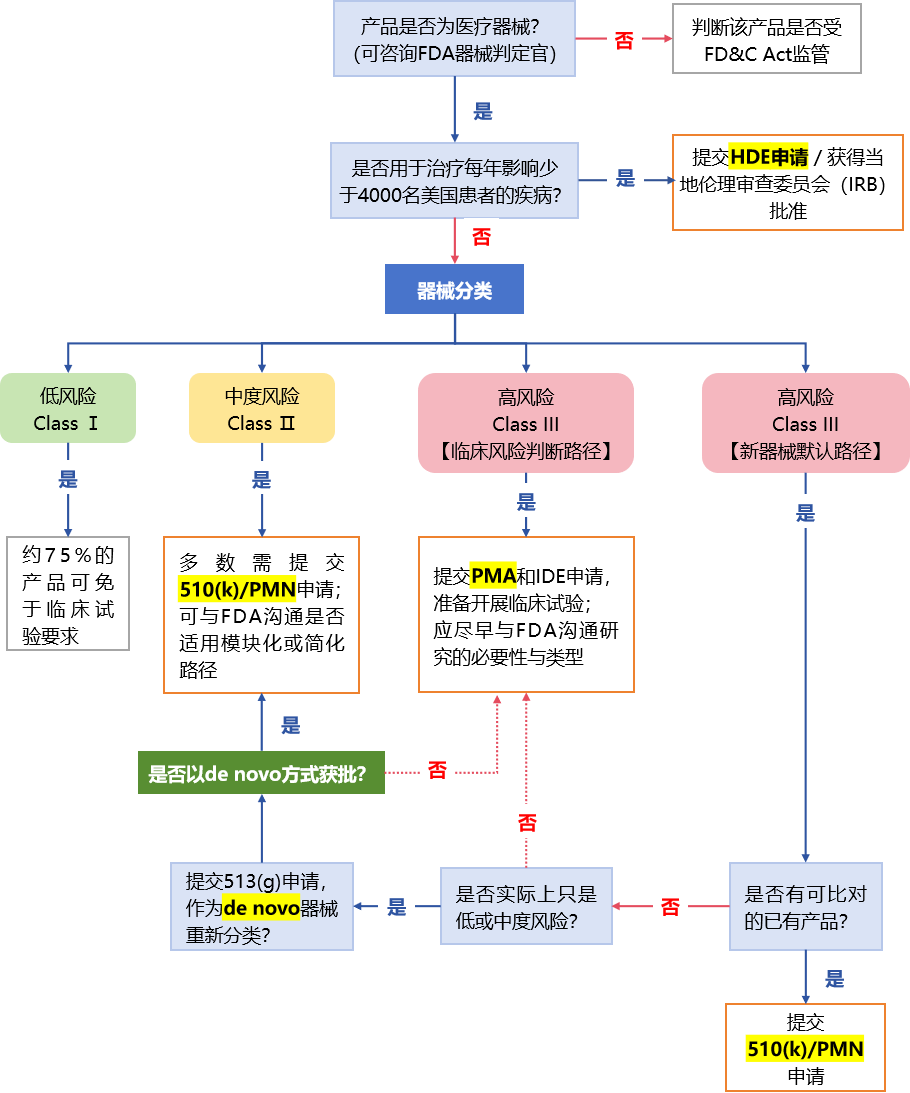
下文将详解PMA、De Nova和510(k)等核心审批路径的申请流程及合规要点,为企业制定FDA上市准入审批策略提供参考。
1.PMA(上市前批准)
上市前批准(PMA)适用于III类(高风险或植入式)及部分高风险的II类医疗器械,是FDA最严格的上市审查程序。申请企业需提供详尽的非临床试验和临床研究数据,以证实产品安全性和有效性,资料一般一次性完整提交(传统PMA),或分模块逐步提交(模块化PMA)。对于罕见病治疗器械,则可通过人道主义使用器械豁免(HDE)程序申报,HDE对有效性证据要求较低,但强调安全性评估。整体来看,PMA流程周期较长(约180天以上),审查严格,企业应做好充分的准备。
2.De Novo(新型医疗器械分类请求)
De Novo程序适用于无市场先例且风险为低到中度的新型医疗器械。这类器械通常无法通过510(k)程序获得实质等同性认可,但风险又未高到必须采用PMA程序。企业通过De Novo申请,可将产品划定为风险较低的I类或II类,从而避免严格的PMA审批。如果FDA接受了De Novo申请,未来类似器械即可通过510(k)路径上市。因此,De Novo是创新型产品进入美国市场的重要通道,申请周期一般为120至150天左右。
3.510(k)(上市前通知)
510(k)适用于具有低到中度风险、市场上已有实质等同产品的医疗器械,通常包括大多数II类和少数I类器械。申请企业需向FDA提交资料,证明新器械与市场上合法销售的等同器械(Predicate Device)具有相同的预期用途和相似的技术特征,或技术差异不影响安全有效性。审批周期一般为90天,企业收到FDA实质等同认可(SE)通知后即可合法上市。510(k)因其流程简化、周期较短,是中国企业最常使用的FDA注册路径。
综上所述,PMA、De Novo和510(k)构成了FDA传统医疗器械注册的三大核心路径。企业应基于产品的风险等级、创新程度以及市场情况综合评估,选择合适的审批路径,以优化合规效率和产品上市进度。
二、基于人工智能/机器学习的医疗器械软件(AI/ML SaMD)的附加考虑因素
(一)相关术语界定
1.医疗器械软件/软件作为医疗器械(Software as a medical device, “SaMD”)
“医疗器械软件”或者“软件作为医疗器械”(SaMD) 这一术语由国际医疗器械监管者论坛(IMDRF)定义为“旨在用于一个或多个医疗目的的软件,且能够在不依赖硬件医疗器械的情况下执行这些医疗目的”,这意味SaMD可以在通用计算平台(如智能手机、平板电脑、个人电脑等)上运行,无需专用的医疗硬件支持。SaMD的使用正在不断增加,可以广泛应用于各种技术平台,包括医疗器械平台、商业现成的平台和虚拟网络等。
值得注意的是,除SaMD以外,与医疗器械相关的软件类型还有两大类:作为医疗器械一部分的软件/嵌入式医疗器械软件(Software in a medical device, “SiMD”)和用于制造或维护医疗器械的软件(software used in the manufacture or maintenance of a medical device)2。FDA在2021年的AI-enabled SaMD指南文件中曾指出,针对SiMD的人工智能应用审批流程,可能与对SaMD的监管要求存在重叠部分,但截至目前,FDA尚未出台专门针对人工智能赋能的SiMD的独立监管指引3。
2.AI/ML赋能医疗器械(Artificial Intelligence and Machine Learning-Enabled Medical Devices)
AI/ML赋能医疗器械是指那些在其设计、开发或功能中融入人工智能和机器学习技术的医疗器械。这些器械能够通过算法分析和处理医疗数据,如影像、传感器数据或患者记录,从而辅助诊断、治疗决策或患者监测。例如,AI/ML赋能医疗器械可以在放射学中自动分析医学影像,识别异常区域;在心脏病学中,实时监测心电图(EKG)数据,检测心律失常;在皮肤科中,分析皮肤病变图像,辅助识别皮肤癌等。这些器械的核心优势在于其持续学习和适应能力,能够随着新数据的积累不断优化性能,从而提高诊断的准确性和治疗的个性化水平。
FDA官网公布了“AI/ML赋能医疗器械清单”,该清单包含所有已获批准的器械介绍。截至2025年8月19日,该清单包含1247款医疗器械,其中16款通过PMA批准,36款获得De Novo批准,其余1195款的审批途径为510(k)4。
(二)FDA应对AI/ML SaMD监管挑战的转型举措
FDA在监管AI/ML SaMD时面临独特挑战:这类软件的自适应进化特性与传统医疗器械监管体系存在根本性冲突。自1976年《医疗器械修正案》实施以来,FDA对已上市医疗器械的重大修改(包括设计变更、组件调整、生产工艺改进或预期用途扩展等可能显著影响设备安全有效性的改动),始终遵循补充申报制度——要求制造商根据初始审批路径(510(k)、De Novo或PMA),提交相应的补充申请。然而,面对具有持续自主优化特征的AI/ML SaMD,这种基于静态产品形态的监管范式已显露出明显的不适应性,显然需要构建全新的监管体系。
为应对这一挑战,FDA采取了一系列监管变革措施,以下是截至2025年1月,FDA发布的与AI/ML SaMD相关监管文件的梳理,涵盖讨论文件、行动计划、指导原则和指南草案等:

可见,FDA于2024年末至2025年初发布了多项指导文件,这些近期文件重点围绕两大主题展开:
一是预定变更控制计划(PCCP),以支持AI系统安全可控地进行性能迭代;
二是全产品生命周期(TPLC)管理,强调从设计开发到后市场监测的全过程监管要求。以下将分别介绍上述举措的核心内容及其对行业合规实践的影响。
1.预定变更控制计划(PCCP)
参考指南:2024年12月——《最终指南:AI赋能器械软件功能的预定变更控制计划上市提交建议》(Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions)
FDA通过引入“预定变更控制计划”(Predetermined Change Control Plan, “PCCP”),以允许制造商提前指定计划的AI/ML修改,从而在保持监管监督的同时实现迭代改进。该指南首先承认,AI赋能器械软件功能(AI-enabled device software functions, “AI-DSF13”)的开发是一个迭代过程,并且其新的PCCP框架旨在提供“合理的安全性和有效性保障”。FDA不再要求AI-DSF制造商在器械合法上市后,对显著的修改进行审批或批准,而是要求AI-DSF制造商在首次市场提交时(即510(k)、De Novo或PMA)包含PCCP。在上市前审查过程中,FDA将审查PCCP,“以确保器械的持续安全性和有效性,而无需为实施PCCP中描述的每个修改提交额外的市场申请”。FDA将不再逐个审查每次修改——这将无法监控和评估——而是审查旨在指导技术持续自我修改的人为定义目标(即算法)。
该指南概述了FDA关于AI-DSF PCCP市场提交内容的建议,通常包括以下几个部分:
- 对具体计划中的器械修改的详细描述(即“修改描述”);
- 用于开发、验证和实施这些修改的相关方法,确保器械在预期使用人群中的持续安全性和有效性(即“修改协议”);以及
- 对计划中的修改及其风险缓解措施的利益和风险评估(即“影响评估”)。
值得注意的是,文件中同时提醒了制造商不应仅依赖该指南来“完整描述可能需要在AI-DSF市场提交中包括的内容”——即FDA小心地澄清,PCCP只是AI-DSF市场提交中的一部分。
指南中概述的PCCP要求是广泛的,既适用于自动AI-DSF修改(即由软件自动实施的修改,也称为“持续学习”),也适用于手动AI-DSF修改(即涉及需要人工输入、操作、审核和/或决策的步骤,因此无法自动实施的修改)。尽管指南中没有明确说明,但可以推测该要求适用于所有未来的市场提交,因为其中没有表明该要求会追溯适用。
2. 全产品生命周期(TPLC)管理
参考指南:《草案指南:AI赋能器械软件功能的生命周期管理和上市提交建议》(Draft Guidance: Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations)
这份草案指南提出了全产品生命周期(Total Products Lifecycle, TPLC)管理的考虑因素以及支持AI-DSF的医疗器械上市提交的具体建议。具体而言,这份草案主要基于FDA此前所批准的1016个AI/ML赋能医疗器械的经验,建议对这类产品采用TPLC方法,以确保其符合安全性和有效性的预期,并详细说明了如何将TPLC方法应用于AI-DSF医疗器械的开发和管理,如何在整个产品生命周期中考虑用户界面、风险应对和数据系统等问题,并提供了向FDA提交文档的示例。
此外,草案指南还包括FDA关于在AI-DSF医疗器械的TPLC过程中解决透明度和偏见问题的最新思路,包括通过收集证据评估设备是否对所有相关人口群体(如种族、民族、性别和年龄)产生类似的益处,从而确保这些设备在预期用途下始终安全有效。
该草案指南提供了针对设备或组合产品中AI-DSF组件的具体建议,但明确指出,FDA关于市场提交的其他指南,包括2023年6月发布的《设备软件功能的上市前提交内容》(Content of Premarket Submissions for Device Software Functions14),仍适用于AI-DSF医疗器械。
FDA指出,该草案指南的公开意见征集期持续至2025年4月7日,最终版的指南将与其他适用的指南一同适用于AI-DSF医疗器械。但值得注意的是,这份草案指南发布于拜登政府执政末期,其后续走向在特朗普政府大幅调整AI政策的背景下存在不确定性。
2025年1月,特朗普总统履职初期即签署行政令,明确撤销了拜登政府的人工智能行政命令第14110号(EO14110,涉及“安全、可信赖 AI”发展原则),并要求各联邦机构审查基于该命令出台的政策和指南。随后,白宫于2025年7月发布《赢得竞赛:美国AI行动计划》(America’s AI Action Plan)及三项配套行政令,强调加速AI创新、减少不必要监管、消除阻碍产业发展的政策负担。在此政策导向下,FDA 等机构可能对先前制定的 AI 相关指南进行重新评估,拜登时期拟定的一些监管举措(如 2025 年 1 月发布的生命周期管理指南草案)可能面临延迟或调整。然而,需要强调的是,截至目前,FDA 并未宣布撤销已发布的 AI 医疗器械指南,而是处于政策审查和潜在修订阶段15。因此,监管走向仍有变数,建议业界持续关注特朗普政府后续的政策落实情况。
三、与HIPAA相关的数据合规问题
除上述FDA的上市审批监管以外,在人工智能医疗器械产品的产品研发和临床实践过程中,往往涉及大量患者的受保护健康信息(Protected Health Information, “PHI”),因此必须同时符合《健康保险可携性与责任法案》(HIPAA)关于隐私和数据安全的要求。
《健康保险可携性与责任法案》(Health Insurance Portability and Accountability Act, “HIPAA”)是美国于1996年颁布的一项联邦法律,旨在保护个人健康信息的隐私和安全。该法案主要适用于医疗保健提供者、健康计划和医疗保健信息交换中心等“涵盖实体”,以及与这些实体合作并处理PHI的“业务伙伴”。作为一部联邦法律,HIPAA授权了美国卫生与公众服务部(Department of Health and Human Services, “HHS”)制定具体的实施细则,通过联邦法规的形式另行颁布独立规则文件,主要包括以下核心规则:
- 隐私规则(Privacy Rule)16:规定了涵盖实体(如医疗服务提供者、健康计划和医疗信息交换中心)在使用和披露PHI时必须遵守的标准,并赋予个人对其健康信息的权利,如查看和请求更正。此外,这一规则也要求涵盖实体采取适当的措施,确保PHI的机密性、完整性和可用性,其主要目标是确保个人健康信息在被保护的同时,允许必要的信息流动,以提供和促进高质量的医疗保健,并保护公众的健康和福祉。
- 安全规则(Security Rule)17:旨在保护以电子形式创建、接收、使用或维护的受保护健康信息(ePHI)的机密性、完整性和可用性。该规则适用于所有涵盖实体以及与这些实体合作并处理ePHI的业务伙伴。安全规则要求涵盖实体采取适当的技术、物理和管理措施,以保护ePHI的机密性、完整性和可用性。此外,安全规则还要求定期进行风险分析和评估,以识别潜在的安全威胁,并采取相应的防范措施。
- 违规通知规则(Breach Notification Rule)18:要求涵盖实体在PHI被未经授权访问或披露时及时通知受影响的个人和相关监管机构。根据这一规则,涵盖实体以及其业务伙伴在发现未经授权访问、获取、使用或披露未加密的PHI的情况下,必须在合理时间内(不超过60天)通知受影响的个人。通知应包括事件的简要描述、涉及的信息类型、受影响个人应采取的保护措施、涵盖实体为调查和缓解事件所采取的措施,以及涵盖实体的联系信息。如果数据泄露影响到500名或以上的州或辖区居民,涵盖实体还必须在60天内通知该州或辖区的主要媒体机构。此外,所有涉及未加密PHI的泄露事件都必须报告给HHS。
- 执法规则(Enforcement Rules)19:执法规则授权HHS下属的民权办公室(OCR)对违反HIPAA隐私、安全和违规通知规则的行为进行调查和处罚,并规定了调查程序、民事罚款的适用范围以及听证程序。具体而言,民事罚款根据违规的严重程度和主观过错程度分为四个等级,单次违规的罚款范围从每次100美元到50,000美元不等,年度累计最高可达150万美元。此外,对于故意违规行为,还可能面临刑事处罚,包括罚款和最高10年的监禁。近年来,随着AI技术的广泛应用和数据泄露事件的增多,OCR加强了对合规风险的监管,通过主动审查、投诉调查和合规审计等方式执行该规则,以促进医疗行业对患者隐私和数据安全的重视。
随着人工智能(AI)在医疗器械领域的广泛应用,确保AI系统在处理PHI时的HIPAA合规性变得尤为重要,包括确保数据的去标识化、与第三方供应商签订业务伙伴协议(Business Associate Agreement, “BAA”)、提升算法的透明度与可解释性,以及强化系统的安全性以防止数据泄露等。在人工智能医疗器械的设计、开发和部署过程中,数据合规性是确保患者隐私和系统安全的关键因素,特别是在远程医疗、云端部署以及跨境数据传输这三个场景中,涉及大量的PHI,面临独特的合规挑战。因此,深入分析这三个场景,有助于识别潜在的合规风险,并制定有效的应对策略。
(一)远程医疗与远程患者监护
在人工智能医疗器械快速发展的背景下,远程医疗(telehealth)和远程患者监控(Remote Patient Monitoring, “RPM”)成为实现个性化医疗和持续护理的重要手段。
具体而言,远程医疗是指通过数字通信技术(如视频会议、移动应用和远程诊断平台)提供医疗服务的模式,涵盖了远程会诊、慢病管理、心理健康支持和健康教育等多种形式,此类技术往往通过可穿戴设备和传感器,持续收集患者的生理数据,如血压、血糖、心率和体重等。这些数据通过无线网络实时传输给医疗提供者,便于医生及时了解患者的健康状况,进行干预和调整治疗方案。
随着技术的进步,AI在远程医疗和RPM中的应用日益广泛。AI算法可以分析大量健康数据,预测疾病风险,辅助诊断,并提供个性化的治疗建议。然而,这类技术在处理PHI时,必须严格遵守HIPAA的规定,以确保患者个人数据和隐私的安全性。具体合规要点包括:
1.最小必要原则(Minimum Necessary Standard)
根据HIPAA隐私规则,涵盖实体在使用、披露或请求PHI时,必须合理限制信息的使用和披露范围,仅限于完成特定任务所必需的信息。因此,涵盖实体在开展系统数据处理活动时,应仅访问和使用完成任务所必需的PHI,避免过度收集数据。
在AI驱动的远程医疗和患者监测中,系统应仅收集与医疗任务直接相关的信息(如糖尿病预测无需患者住址),并通过去标识化技术降低隐私风险。同时,应严格限制数据访问权限,例如医生可查看完整病历,而技术人员仅接触匿名化数据,同时采用加密和多因素认证保障安全。其次,在数据处理时,应优先通过分散计算或模拟数据训练AI模型,避免集中存储敏感信息。此外,需定期评估数据需求、审计系统漏洞,并更新防护措施以应对新风险。最后,医护人员和技术人员应接受针对性培训,确保日常操作符合规范,并建立违规响应机制及时处理问题。
2.数据去标识化(De-identification)
根据HIPAA隐私规则,涵盖实体在使用AI模型进行训练时,需确保数据已根据HIPAA的“专家判定”(Expert Determination)或“安全港”(Safe Harbor)标准进行去标识化,并防范数据再识别的风险。
专家判定法要求由具备统计和科学知识的专家评估数据的可识别性。专家需应用公认的统计和科学原则,确定信息被预期接收者单独或结合其他合理可得信息用于识别个体的风险非常低,并记录分析方法和结果以证明该判断20。
安全港法则规定了一个更为具体的操作流程,要求从数据集中删除18种特定的标识符,例如姓名、地理位置(州以下的细分)、所有与个人直接相关的日期(仅保留年份)、电话号码、社会安全号码、医疗记录号码等。此外,涵盖实体必须没有实际证据表明剩余信息可以单独或与其他信息结合用于识别个体21。
在AI技术处理复杂自由文本医疗数据时,这两种方法各有优劣:专家判定法提供了更大的灵活性,适用于需要在保留数据实用性的同时确保隐私的复杂情境,而安全港法则因其明确的删除标准而易于实施,适合处理大规模数据集。因此,相关制造商在选择去标识化方法时应根据具体的应用场景、数据特性以及对数据适用性和隐私保护的需求权衡决定。
3.患者知情同意
虽然HIPAA并未对知情同意的具体形式和内容作出详尽规定,但其隐私规则要求涵盖实体在使用或披露PHI之前,必须获得患者的授权,除非该使用或披露属于治疗、支付或医疗保健运营等例外情况。
在远程医疗和RPM场景中,AI医疗器械会收集、分析和传输患者的健康数据,因此,制造商一方面应告知患者其健康数据将如何被收集、存储和使用,尤其是在AI系统中用于诊断、治疗或其他医疗决策支持的情况;另一方面,也应当详细说明患者的健康数据可能会与哪些第三方共享;同时,制造商也应告知患者在HIPAA下所享有的权利,包括访问其健康信息、请求更正、限制某些信息的使用或披露,以及提出投诉的途径。此外,相关涵盖实体应当注意根据美国各州的具体法规,制定适当的知情同意流程和文档记录。
(二)云端部署
云端部署是指将软件应用、服务或数据托管在远程的云计算平台上,而非传统的本地服务器。这种方式利用云平台的弹性计算资源和分布式架构,实现按需扩展、灵活配置和高可用性。在医疗领域,云端部署特别适用于处理大规模的医疗数据和复杂的AI模型,不仅可以加速模型的训练和迭代,还能实现跨地域的数据共享和远程协作。例如,微软的Cloud for Healthcare平台提供了专为医疗保健组织设计的基于云的解决方案,支持数据的集中管理和分析22。
然而,AI医疗器械的云端部署也带来了数据安全和隐私保护的挑战。为确保HIPAA合规,需注意以下方面:
1.技术保障措施(Technical Safeguards)
根据HIPAA安全规则,涵盖实体必须实施技术保障措施以保护ePHI,包括访问控制、审计控制、完整性控制和传输安全,这些措施共同构建了ePHI的安全防护体系。
- 访问控制:HIPAA要求涵盖实体必须实施技术策略和程序,以确保只允许授权人员访问 ePHI。医疗器械制造商应实施细粒度的访问控制策略,确保只有授权人员才能访问ePHI。这包括为每位用户分配唯一的用户ID,并结合多因素身份验证(MFA)机制,增强身份验证的安全性。例如,Microsoft Entra ID提供了基于角色的访问控制和条件访问策略,帮助有效管理用户权限23。
- 审计控制:HIPAA要求所涉实体必须实现硬件、软件和/或程序机制,记录和检查包含或使用ePHI的信息系统中的活动。因此,AI医疗器械制造商应考虑在应用系统中实现对访问或操作ePHI活动记录的功能。譬如,可通过云日志的方式对所有访问或使用云资源的活动日志进行实时查询和转储,并作出实时决策分析。
- 完整性控制:HIPAA要求所涉实体必须执行一个策略和程序,防止ePHI被篡改或破坏,并实施电子机制,以确认ePHI未被非授权地更改或销毁。AI医疗器械制造商应考虑采用行业任何的加密算法对数据进行保护,比如采用云端部署第三方的数据加密服务,通过分层密钥管理机制,方便各层密钥的轮换,防止信息系统被入侵或篡改。
- 传输安全性:HIPAA要求所涉实体必须实现技术安全措施,以确保网络传输中的ePHI免受非授权访问。因此,在数据传输和存储过程中,采用强加密技术是保护ePHI的关键措施,包括使用安全传输层协议(TLS)加密数据传输,以及对存储在数据库中的ePHI进行加密处理。
2.业务伙伴协议(BAA)
根据HIPAA规定,任何代表受涵盖实体处理、存储或传输PHI的第三方,均被视为业务伙伴,必须签署BAA24。即使云服务提供商无法解密存储的数据,只要其处理PHI,也必须承担相应的合规责任,因此AI医疗器械的开发商在选择云服务提供方时,必须确保与所有处理PHI的第三方服务商签订BAA,以明确双方在数据保护方面的责任。
具体而言,BAA的核心内容包括:
- 数据使用和披露范围:明确业务伙伴在处理PHI时的权限和限制,确保其用途仅限于合同规定的服务范围;
- 安全保障措施:要求业务伙伴实施适当的行政、物理和技术安全措施,以保护PHI的机密性、完整性和可用性。
- 数据泄露通知义务:规定在发生数据泄露事件时,业务伙伴必须在规定时间内通知受涵盖实体,以便及时采取应对措施。
- 合同终止后的数据处理:在合同终止时,业务伙伴应返还或销毁所有PHI,除非返还或销毁不切实际,此时应继续保护PHI并限制其使用。
(三)跨境数据传输
HIPAA的适用范围取决于PHI的来源和处理实体,而非数据所在地域,即只要由美国的医疗提供者(涵盖实体)委托境外第三方处理PHI,则该境外实体就成为HIPAA下的业务伙伴,必须遵守与美国本土同等的安全与隐私保护要求25。在此背景下,AI医疗器械开发企业需要全面了解并落实HIPAA对于跨境数据传输的合规要点,确保无论数据是从美国流向境外,还是境外访问美国云端,都符合法规要求,保障患者隐私安全。跨境传输患者数据在带来便利的同时,也面临一系列独有的合规挑战:
首先是安全风险增高与法律管辖难题。虽然HIPAA并不禁止将电子PHI存储或处理在境外服务器上,但境外环境的复杂性可能导致PHI面临更大的风险和脆弱性(如某些特定国家的黑客攻击或恶意软件攻击企图显著更频繁)。同时,在数据出境发生违规时,监管部门追责和执行处罚会更复杂——这要求企业在进行安全风险评估时,将境外存储和传输的额外风险纳入考量。
其次是合规标准差异与人员意识不足。境外合作方所在国家/地区的隐私法规和安全标准可能与HIPAA存在不一致的规定,医疗数据跨境流动需要同步遵守多国法规:例如在远程医疗中既要符合美国HIPAA,也要考虑患者和医生所在国的隐私法律要求。此外,如果境外参与处理PHI的员工或技术人员缺乏HIPAA培训,可能因为意识不足而发生违例操作——如未获授权查看患者记录、未及时退出包含PHI的系统或在遇到潜在数据泄露时处理不当。这些都增加了跨境协作的合规难度,要求对境外人员进行同样严格的背景审查、培训教育和定期合规审计。
最后,技术和基础设施差异也是一大挑战。不同国家的数据中心和IT基础设施在物理和技术防护上可能存在差异,远程医疗大量依赖于云平台和网络传输,如果境外网络环境质量不佳或设备技术落后,也可能影响数据传输的可靠性和安全性。因此,跨境医疗数据管理需要更高强度的尽职调查和防护规划——对境外合作伙伴的审查和风险管控应不亚于对国内伙伴的要求,必要时甚至更严格。
综上所述,跨境数据传输已成为医疗科技创新和服务模式中不可避免的一环。远程医疗和云端部署等应用虽突破了地域限制,但也要求AI医疗器械企业以更高的标准来保护患者隐私。HIPAA在数据传输、访问控制、加密、防护以及业务伙伴管理方面为跨境数据流动设定了清晰的要求和原则。企业只有充分识别跨境环境下的独特风险,并将HIPAA合规要点融入日常运营(包括签订严格的BAA、采用领先的加密传输技术、强化访问管理和人员培训等),才能在拓展国际业务的同时确保患者健康信息始终受到应有的保护。
四、结语
随着AI技术在医疗器械领域的深入融合,美国FDA与相关监管机构正持续推进监管框架的更新,以更好地回应技术迭代带来的挑战。本文从传统审批路径入手,结合AI/ML赋能器械的软件属性、数据动态性以及在数据安全与隐私保护方面的特殊需求,分析了近年来美国最具代表性的监管变革路径。对于希望进入美国市场的中国医疗器械企业而言,理解PMA/510(k)等核心审批机制仍是合规起点,而对PCCP、TPLC、HIPAA等新兴合规要求的及时响应,将成为企业提升产品信任度与加快全球布局的关键。下一篇文章将转向欧盟AI医疗器械的合规视角,进一步补足全球合规图景。
注释:
1See: Artificial Intelligence Index Report 2025, https://hai.stanford.edu/ai-index
2See:https://www.fda.gov/medical-devices/digital-health-center-excellence/software-medical-device-samd
5See:Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) - Discussion Paper and Request for Feedback, https://www.fda.gov/media/122535/download?attachment
6See:AI/ML SaMD Action Plan, https://www.fda.gov/media/145022/download?attachment
7See: Good Machine Learning Practice for Medical Device Development: Guiding Principles, https://www.fda.gov/medical-devices/software-medical-device-samd/good-machine-learning-practice-medical-device-development-guiding-principles
8See: Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions, https://www.fda.gov/media/166704/download
9See: Predetermined Change Control Plans for Machine Learning-Enabled Medical Devices: Guiding Principles, https://www.fda.gov/medical-devices/software-medical-device-samd/predetermined-change-control-plans-machine-learning-enabled-medical-devices-guiding-principles
10See: Transparency for Machine Learning-Enabled Medical Devices: Guiding Principles, https://www.fda.gov/medical-devices/software-medical-device-samd/transparency-machine-learning-enabled-medical-devices-guiding-principles
11See: Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/marketing-submission-recommendations-predetermined-change-control-plan-artificial-intelligence
12See: Draft Guidance: Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/artificial-intelligence-enabled-device-software-functions-lifecycle-management-and-marketing
13FDA将AI-DSF定义为:“一种设备软件功能,它实现一个或多个AI模型。”
14See: Content of Premarket Submissions for Device Software Functions, https://www.fda.gov/media/153781/download
15See: FDA Issues Draft Guidance for AI-Enabled Devices: Key Takeaways, https://www.dentons.com/en/insights/alerts/2025/february/11/fda-issues-draft-guidance-for-ai-enabled-devices
16See:https://www.hhs.gov/hipaa/for-professionals/privacy/laws-regulations/index.html
17See:https://www.hhs.gov/hipaa/for-professionals/security/index.html
18See:https://www.hhs.gov/hipaa/for-professionals/breach-notification/index.html
19See:https://www.hhs.gov/hipaa/for-professionals/special-topics/enforcement-rule/index.html
20See:https://www.private-ai.com/en/blog/hipaa-expert-determination
21See:https://www.private-ai.com/en/blog/hipaa-compliance-private-ai
22See:https://learn.microsoft.com/zh-cn/industry/well-architected/healthcare/architecture-overview
(原标题:卓纬研究 | AI医疗器械全球合规系列(上篇)——美国篇:FDA和HHS的监管路径与合规解析)
(来源:卓纬律师事务所,作者:李诘、方云竹。本文已获作者授权转载。)
特别声明:本微信公众号的文章仅供交流之用,不代表北京卓纬律师事务所或其律师的正式法律意见或建议。


