





引言
人工智能技术正以前所未有的速度渗透医疗器械领域,从影像诊断、疾病预测,到个体化治疗与闭环控制,AI已成为推动全球医疗科技变革的核心动力。随着算法从辅助工具升级为影响临床决策的关键组件,监管体系的演化也相应进入深水区——如何在促进创新与确保安全之间找到平衡,逐渐成为各国监管框架的核心议题。
在全球范围内,各国监管机构正陆续提出和探索AI/ML医疗器械的监管路径,但欧盟仍以其独特的“双轨式监管模式”走在前列。欧盟并未将人工智能医疗器械(Medical Device Artificial Intelligence, “MDAI”)视为单一监管对象,而是采用横纵叠加的体系式治理方式:医疗器械安全与性能要求由MDR/IVDR构建,算法风险治理与基本权利保护由AIA赋能,两套体系在欧盟新立法框架(NLF)的原则下协同运作,形成全球最系统化的MDAI监管框架。
在此背景下,理解欧盟MDAI监管体系的边界、逻辑与合规要求,已成为所有希望进入欧盟市场的创新型医疗企业的前提。本文旨在以法规之间相互作用关系为切入点,从MDR/IVDR的基础逻辑、AIA的横向规制、MDAI的界定与风险认定机制,到技术文件、数据治理、人类监督及上市后监测等具体合规要求,系统梳理MDAI在欧盟的完整合规路径,并提出面向中国企业的实践建议。
一、MDR与IVDR的监管核心逻辑
在欧盟的监管体系中,医疗器械(Medical Devices, “MD”)与体外诊断医疗器械(In Vitro Diagnostic Medical Devices, “IVD”)分别受到两部并行法规的约束:《医疗器械法规》(The regulation on medical devices, Regulation (EU) 2017/745, “MDR”)与《体外诊断医疗器械法规》(The Regulation on In Vitro Diagnostic Medical Devices, Regulation (EU) 2017/746, “IVDR”)。这两部法规共同取代了早期的指令体系(MDD1与IVDD2),构成了欧盟医疗器械监管的纵向基础。对于人工智能驱动的医疗产品而言,理解MDR与IVDR的基本逻辑,是把握后续欧盟《人工智能法案》(Artificial Intelligence Act, “AIA”)横向监管的前提。
(一)医疗器械与体外诊断的监管边界
MD与IVD的分野,首先源自器械用途的不同:
- MD是指用于人体内部或直接作用于人体的医疗器械,包括诊断、治疗、监测、补偿或替代生理功能的设备、软件及配件。
- IVD是指用于体外检测人体样本(如血液、尿液、组织)的器械、试剂或软件,其目的是提供医学信息以支持诊断或疾病预测。
MDR与IVDR二者的立法理念相似,均强调“安全性、性能、可追溯性”三大支柱,但监管重心不同:MDR更注重患者直接安全与临床有效性,IVDR则更强调科学有效性、分析性能与临床性能的链式验证。
在AI医疗产品的语境中,这一区别并非理论化的。例如,一个基于AI算法的心电信号诊断软件属于MDR监管;而一个AI算法用于基因测序结果解读的系统,则落入IVDR管辖范围。因此,在进行AI产品的“资格判断”(qualification)时,首先应确定其最终用途:是作用于人体本身还是用于体外样本分析——这一点决定了不同法规的适用选择路径。
(二)风险分类机制与公告机构(NB)角色
MDR与IVDR都采用基于风险的监管模式,即产品风险等级越高,合规要求越严:
- MDR将器械分为四级:I、IIa、IIb、III(附录 VIII)。分类规则综合考虑器械的侵入性、用途、持续时间及对患者的潜在影响。
- IVDR则依据附录VIII将产品划分为A、B、C、D四级,评价维度更侧重公共卫生风险与诊断关键性。例如,涉及高危传染病检测的试剂(如HIV、乙肝检测)属于 Class D;肿瘤标志物或遗传检测等多数AI驱动诊断算法则多落在Class C。
在合格评定路径上,两部新法规均引入了“公告机构”(Notified Body, “NB”)作为独立的技术审查方。具体而言,NB并不是主管机构的一部分,而是独立的组织,由欧洲经济区成员国的主管机构指定,负责在欧盟法规要求第三方合格评定的情况下,对需加贴CE标志的产品进行符合性评估。对于MD和IVD而言,NB的法律定义为“根据本法规指定的合格评定机构”(a conformity assessment body designated in accordance with this Regulation)3。
在旧指令时代(MDD/IVDD),大部分IVD可由制造商自我声明合格,但在新法规下,这一自我认证时代已经终结4。
- 根据MDR,大部分中高风险器械(IIa及以上)需经NB审查;
- 根据IVDR,除最低风险的Class A非无菌产品外,几乎所有IVD都需要NB介入。
这种体系变化直接导致了欧盟NB资源的紧张,也推动了企业必须更早介入NB沟通。对于MDAI而言,NB不仅关注传统安全与性能,还会审查算法训练数据、模型验证、软件生命周期管理及可解释性等内容。
(三)MDR与IVDR的对比
MDR和IVDR是两套独立但互补的法规体系,各自设定了阈值和清单,适用于不同类型的产品。以下是两者的主要区别:

二、相关医疗器械的判定与应用场景
(一)软件作为医疗器械(SaMD/MDSW)
随着软件在诊断与治疗中的普遍使用,欧盟监管体系对“软件作为医疗器械”(Software as a Medical Device, “SaMD”)或“医疗器械软件”(Medical Device Software, “MDSW”)建立了独立规则体系。
MDR第2条明确规定,若软件的预期用途在于“由制造商意图用于提供诊断或治疗决策信息”,则被视为医疗器械。欧盟医疗器械协调小组(Medical Device Coordination Group,“MDCG”)在其发布的指南MDCG 2019-115中,进一步细化了判断标准。该指南要求制造商结合以下五个步骤进行判断:
- 确认产品是否符合指南对“软件”的定义,若其不属于软件范畴,则不适用软件指南。
- 软件是否作为MDR附录XVI产品、医疗器械的组成部分或附件,或是否驱动或影响医疗器械的使用;若答案为是,则直接被纳入MDR/IVDR监管。
- 评估软件是否执行超出单纯存储、归档、通信、简单检索或无损压缩的操作,例如是否对医学信息进行分析、计算或解释;若执行此类操作,则可能构成MDSW。
- 判断软件的输出是否旨在服务“个体患者”的临床决策,例如生成诊断、监测或治疗所需的决策触发信息;若仅用于群体数据汇总、科研或流行病学用途,则不属于医疗器械软件。
- 对前述步骤进行综合判断,以确认该软件是否满足指南对医疗器械软件的定义,从而决定其是否应遵循MDR/IVDR下的风险分类与CE合格评定程序。
因此,软件是否属于医疗器械,核心仍在于其输出是否以医疗目的服务于个体患者的临床决策。若AI软件仅用于教学研究、行政管理、信息检索或通信功能(如PACS系统、医院排班软件),则不构成医疗器械;若其分析结果被医生用于诊断、风险预测或治疗决策,则必须受MDR或IVDR的调整。
例如:
- 利用深度学习模型分析 CT 影像识别肺结节的系统——适用MDR;
- 基于算法对血液检测结果进行定量解释的系统——适用IVDR;
- 用于医院工作量管理或统计分析的AI工具——通常不属于医疗器械。
值得注意的是,欧盟法规强调“意图用途(intended purpose)”的表述权在制造商,但监管机构会审查说明书、用户界面及算法输出内容,以判断其真实功能。
(二)人工智能医疗器械(MDAI)
人工智能医疗器械(Medical Device Artificial Intelligence, “MDAI”)在欧盟法规体系中并非一个独立的法律分类,而是欧盟人工智能委员会(Artificial Intelligence Board, “AIB”)和MDCG为说明AIA与医疗器械法规适用关系而创设的工作性概念。
如前所述,现行MDR和IVDR通过对“软件作为医疗器械(MDSW/SaMD)”的资格与风险管理要求,已经一定程度上覆盖了与软件相关的风险,但仍缺乏对AI系统独有风险的规制6。为弥补这一空白,AIB和MDCG在相关指南中,从AIA引入“AI system”的定义,并结合MDR/IVDR所界定的医疗器械范围,将这一交叉集合中的人工智能系统统称为人工智能医疗器械(MDAI)。
综上,MDAI是指在MDR/IVDR监管框架内使用且符合AIA所定义的AI system的人工智能系统。此类系统既需满足MDR/IVDR对在临床、安全与性能方面的要求,也需遵循AIA 对高风险AI系统提出的数据治理、透明度、人类监督与风险管理等附加义务。MDAI的概念界定旨在统一描述这一“人工智能作为医疗器械使用”的交叉领域,明确在欧盟监管体系中其双重合规属性。
三、AIA的横向监管:对高风险MDAI的要求
在MDR与IVDR构建的纵向监管体系之上,欧盟于2024年正式通过欧盟《人工智能法案》(Artificial Intelligence Act, “AIA”),确立了全球首个全面的人工智能立法框架。这部法规的出台,标志着MDAI的监管进入“纵横叠加”的新阶段——企业不仅要符合医疗器械法规的安全与性能要求,还要满足AIA对高风险AI系统提出的治理与透明度义务。
(一)AIA的定位及与MDR/IVDR的协调适用
AIA作为一部横向基础法,适用于所有在欧盟市场上提供或使用的AI系统,无论其提供者是否位于欧盟境内,其核心目标在于确保AI系统的安全性、透明性与可解释性,防止算法带来的歧视与偏倚,AIA的制定标志着欧盟在人工智能治理层面迈入了系统性与风险导向的新阶段。
如前所述,尽管MDR与IVDR的要求已对SaMD/MDSW的风险做出了详尽规定,但二者并未针对MDAI所特有的算法风险、动态学习特征及数据偏倚等问题建立明确的监管框架。AIA的引入弥补了这一监管空白:其通过确立对AI系统在健康、安全及基本权利方面潜在危害的识别与防控合规要求,从而对MDR/IVDR进行补充。
根据欧盟新立法框架(New Legislative Framework, “NLF”)原则7,对于包含一个或多个高风险人工智能系统的医疗器械产品,MDR/IVDR与AIA应当并行适用、相互补充。
对此,AIA在第8条第2款关于“对高风险AI系统的合规要求”的条文中明确:
“如果产品包含一个人工智能系统,而本条例的要求以及附件一A节所列欧盟统一立法的要求适用于该系统,则提供者应负责确保其产品完全符合适用的欧盟统一立法的所有适用要求。在确保第1段中提及的高风险人工智能系统符合本节规定的要求时,为了确保一致性、避免重复和最小化额外负担,提供者可选择酌情将其提供的有关其产品的必要测试和报告流程、信息和文件整合到附件一A节所列欧盟统一立法要求的已有文件和程序中。”
欧盟相关监管指导文件也指出:
“为确保一致性、避免重复并最大限度地减少额外负担,MDAI制造商,根据AIA第8条第2款的规定,有权选择在适当情况下将其针对MDAI所提供的必要测试与报告流程、信息及文件,整合入其已根据MDR/IVDR建立的文件与程序之中。强烈鼓励MDAI制造商利用AIA第8条第2款所提供的灵活性。然而,在适用该项灵活性时,MDAI制造商应确保其MDAI完全符合AIA、MDR或IVDR及任何其他适用之欧盟立法的所有适用要求。”
(二)AIA第6条:高风险AI系统的耦合式判定机制
AIA第6条明确了一个AI系统是否属于高风险AI系统(High-Risk AI System)的判断规则。该条文并未简单列举行业或产品,而是通过一种耦合式判定机制,即将AI系统的技术特征与既有欧盟产品安全立法(如MDR/IVDR)相衔接,从而实现风险分级的自动识别。
梳理其立法逻辑如下:

具体到医疗器械领域,AIA的附件一规定了“欧盟统一立法清单”,其中A节第11段和第12段明确包括了MDR(Regulation (EU) 2017/745)和IVDR(Regulation (EU) 2017/746)。因此,凡是“作为或嵌入医疗器械或体外诊断医疗器械”而运行的AI系统,一般均满足AIA第6条第一步所要求的“功能属性”条件。关于第二步的“监管属性”,即该产品是否需要经过NB进行第三方合格评定,则取决于其在MDR与IVDR下的风险等级分类。不同风险等级对应不同的审查要求,也由此决定该AI是否被最终认定为“高风险AI系统”。

因此,在MDR和IVDR框架下,几乎所有中高风险等级的医疗器械所配备或嵌入的AI系统均会自动触发高风险机制,仅极少数可由自我声明合格的低风险产品可享受豁免。换言之,凡医疗器械或体外诊断器械一旦需要NB参与合格评定,其所嵌入的AI模块即自动被视为AIA意义下的“高风险AI系统”,须同时遵守MDR/IVDR与AIA的双重监管。
对于制造商而言,这意味着:无需再单独判断MDAI是否“高风险”,但必须在质量管理体系和技术文档中系统地整合AIA对高风险AI系统所要求的合规义务。
四、高风险MDAI的核心义务
以AIA第8至15条为逻辑主线,将高风险MDAI的要求整合嵌入MDR/IVDR监管架构之中,主要形成了以下四个层面的合规映射:
(一)管理体系(Management Systems)
高风险MDAI的监管体系在MDR/IVDR与AIA下形成互补格局,共同确保系统全生命周期的安全性、性能一致性与对基本权利的保护。
首先,在生命周期管理方面,MDR与IVDR要求制造商在整个产品生命周期内建立持续的监测与管理机制;AIA进一步强化了这一要求,明确高风险AI系统需在设计、开发、测试、部署及更新的各阶段接受系统性审查与监督,建立可持续运行的生命周期管理体系。特别是针对具备持续学习能力的MDAI,AIA规定应通过上市后监测体系实现动态风险识别与性能一致性维护,并确保必要信息传达至部署者、患者及监管机构。
其次,在质量管理体系(Quality Management System, “QMS”)方面,三部法规均要求制造商建立系统化、文件化并持续改进的质量管理体系,以确保符合法规与技术要求。在传统医疗器械监管实践中,MDR/IVDR虽未对QMS的技术性细节作出明确规定,但通常通过引用协调技术标准实现合规落地,其中ISO 1348511作为医疗器械QMS国际通行标准,已成为NB在开展符合性评估时普遍采用的基础框架。AIA第17条明确提供者须建立涵盖风险管理、数据治理、上市后监测及事件报告等要素的QMS,并允许其与MDR/IVDR下的体系进行整合,以减少重复并保持一致性。此外,针对高风险MDAI的质量管理体系的欧洲及国际协调标准的制定工作也正在同步进行中。
最后,在风险管理体系方面,AIA、MDR与IVDR均要求制造商降低已识别与评估的风险,并在其风险管理体系中处理可合理减轻的风险。AIA强调通过全面的风险评估、风险缓解措施的文件记录以及对系统性能的持续监测,确保高风险MDAI在医疗环境中的安全性与可靠性。高风险MDAI的制造商可将AIA第9条中针对MDAI规定的附加风险管理要求,连同AIA规定的测试、报告流程及相关信息与文件,整合入其依据MDR及IVDR已建立的风险管理与程序之中。
(二)数据治理(Data Governance)
AIA与MDR/IVDR均对MDAI的数据治理进行了规制,但其适用范围与重点有所不同:MDR与IVDR着眼于医疗器械临床数据的科学性与有效性,而AIA则关注人工智能系统中数据的质量、代表性与偏差防控,以确保算法运行的安全与合规。
首先,MDR与IVDR确立了基于临床证据的数据治理要求。根据MDR/IVDR的规定,用于器械评估的临床数据必须稳健(robust)、可靠(reliable),并且来源于设计良好的研究(derived from well-designed studies)。为支持充分临床证据的生成,临床评估(clinical evaluation, 适用于MD)或性能评估(performance evaluation, 适用于IVD)必须基于与器械预期用途相符的临床数据。用于验证、确认及控制器械设计的程序与技术,以及由此产生的文件化信息,均应作为临床证据的一部分予以记录,并由NB进行审查。
其次,AIA第10条在前述基础上进一步构建了针对高风险AI系统的数据治理框架。该条规定,用于训练、验证与测试MDAI的数据集应具备相关性、充分代表性,并在最大可能范围内无错误、完整且具有适当统计特征。制造商需建立系统化的数据管理程序,涵盖数据获取、分析、标注、存储、保护及再利用等环节,确保数据在整个生命周期内的完整性与透明性。AIA同时要求制造商识别和减轻可能影响健康、安全或基本权利的偏差,防止由算法或数据歧视引发的不平等结果。
此外,AIA还通过记录保存与日志记录机制强化数据治理的可追溯性。高风险MDAI应具备在整个生命周期内自动记录事件(logs)的能力,以识别因数据偏差或系统更新导致的潜在风险,并作为上市后监测体系的重要组成部分。该要求与MDR/IVDR对临床数据持续监测的义务相衔接,形成闭环监管。
最后,在涉及个人数据时,AIA要求制造商的数据治理措施应完全符合《通用数据保护条例》(GDPR),并对数据收集目的、处理方式及合法性保持透明。制造商可通过第三方认证服务验证数据治理的合规性,从而提升监管信任与审查效率。
综上,AIA侧重算法层面的数据质量与偏差控制,MDR与IVDR则聚焦临床层面的数据可靠性与性能验证,共同确立了高风险MDAI的数据治理核心义务,即:高质量与代表性数据的使用、系统化的数据治理与偏差防控机制,以及全生命周期的数据可追溯与透明合规要求。
(三)透明度与人类监督(Transparency & Human Oversight)
1、透明度义务
在欧盟监管框架中,AIA与MDR/IVDR均将透明度视为高风险MDAI的基础性义务,并通过相互补充的机制在系统设计、部署与使用各阶段实现信息可获得性与可理解性。
首先,AIA通过多条条款确立透明度的制度结构。
一方面,AIA第13条要求提供者确保系统运行具有“足够透明度”,并提供清晰、易懂的使用说明,以便部署者作出知情决策。这包括说明系统的预期用途、禁止用途、功能边界以及人工智能组件对结论生成过程的贡献。此外,对于涉及直接与自然人交互的系统场景下,AIA第50条特别要求用户明确被告知正在与AI互动,除非该事实对合理用户而言已可明显识别。
另一方面,AIA第10条规定了高风险AI系统的数据治理义务,要求训练、验证与测试数据具备相关性、代表性、完整性并在最大可能范围内无偏差。该条亦要求制造商建立数据管理与文件化流程,使监管机构和部署者能够理解AI性能的基础数据结构。进一步地,AIA第12条要求高风险系统保持充分的日志记录,以支持“功能可追溯性”(functional traceability),便于上市后监督与责任落实。
其次,MDR与IVDR通过附件I《通用安全和性能要求》(General Safety and Performance Requirements, “GSPRs”)强化透明度义务。
GSPR一方面要求制造商提供关于器械预期用途、运行方式、风险及限制的充分信息,包括软件(含AI组件)对器械性能的贡献方式,另一方面则要求软件设计遵循当前技术水平,并纳入生命周期管理及风险管理流程,从制度上确保可解释性与文件可追溯性。此外,根据MDR/IVDR附件II和附件III,制造商须在技术文件中详细说明AI的设计逻辑与运行方式、输入数据处理方式及输出生成机制,使部署者和NB能够理解AI对器械风险特征的影响。
通过上述机制,MDR/IVDR与AIA协同确保NB、部署者与用户能够理解高风险MDAI的性质、运行方式及其局限性,支持知情使用与监管审查。
2、人类监督义务
在人类监督方面,AIA与MDR/IVDR共同确立了以“人工最终控制”为核心的安全保障框架,要求高风险MDAI在设计、开发与使用全流程中保持可控性,使人工操作人员始终能够理解、监测并在必要时干预或覆盖系统决策。
根据AIA第14条第1款,高风险MDAI的设计与开发必须内置适当的人类监督机制,使人工操作人员能够理解、监测并在必要时干预系统行为;第2款进一步确认,人类监督亦构成风险管理中的缓解措施。AIA在序言部分也要求制造商应确保系统预设的操作约束不得被系统自身覆盖,且系统必须能够对人工操作指令作出响应。这意味着高风险MDAI在关键决策过程中必须始终保留对人类干预的开放性,确保人工判断仍为最终控制来源。
在MDR/IVDR框架下,主要通过要求MDAI的设计应达到一定的GSPR来支持人类监督原则。为确保高风险MDAI的安全和有效使用及部署,并使医务人员及医疗机构能够实施适当监督,法规要求将监督机制、操作限制及干预方式在使用说明、培训材料及技术文件中明确文件化,使人工监督具备可操作性并融入器械的安全与性能保障体系。
(四)准确性、稳健性与网络安全(Accuracy, Robustness and Cybersecurity)
MDR、IVDR及AIA三部法规均确立了统一的监管原则,即高风险MDAI在上市前与上市后阶段,必须具备稳健的网络安全措施,以确保在普遍认可的技术水平下实现对公众健康和安全的高水平保护。监管框架明确指出,任何与器械运行相关的风险都必须控制在可接受范围内,这要求制造商在器械所有可能的运行模式下,建立效益与风险之间的适当平衡。为此,制造商必须超越单一的功能安全视角,综合考量“器械安全”(safety)与“网络安全防护”(security)之间的相互依存关系,确保两者在风险控制中实现协同。
针对MDR、IVDR及AIA的具体规定,制造商需从技术实现、数据资产保护及管理体系融合三个维度履行此项义务。
首先,在技术防护层面,制造商实施的网络安全措施应具有针对性,旨在防止未经授权的访问、网络攻击、系统利用与数据篡改,并确保系统的运行韧性。特别是AIA要求高风险MDAI必须采取特定的技术手段,以应对人工智能特有的脆弱性22。这种保护不仅涵盖系统本身,还须延伸至人工智能特定资产(如训练数据集、已训练模型)以及底层的信通技术(ICT)基础设施23。其次,在数据治理层面,制造商应采取措施保障数据传输与存储的安全,防止数据与模型投毒,并具备检测与应对网络安全事件的能力。这些要求贯穿于MDAI开发的全过程,包括在训练、验证和测试阶段落实相应的安全防护措施。最后,在管理体系层面,鉴于网络安全是高风险人工智能系统的基本要求及制造商的法定义务,相关措施必须纳入风险管理体系和质量管理体系。制造商需开展风险评估以识别漏洞并实施缓解措施24,且上述体系均须接受合格评定程序的审查25。
五、合规路线与中国企业的应对
(一)MDAI合规地图:从开发到上市后的全流程管理
在前文对高风险MDAI的四类核心合规义务进行了系统阐述后,可以看出,这些要求贯穿产品从设计开发到实际使用的全生命周期。为清晰呈现这些要求在开发、上市前和上市后阶段的具体分布及衔接,以下将以“合规地图”的形式总结MDAI的全流程监管要求、关键活动和整体合规框架:


(二)经济运营方与在欧角色配置
根据MDR/IVDR与AIA的统一框架,向欧盟投放MDAI产品的中国企业必须建立完整的经济运营方体系,包括制造商(Manufacturer)、欧盟授权代表(Authorised Representative, “AR”)、进口商(Importer)和分销商(Distributor),以及合规负责人(Person responsible for regulatory compliance, “PRRC”)。
制造商是法律上承担全部合规义务的主体,需确保产品符合MDR/IVDR与AIA所要求的安全性、性能、数据治理、透明度、人类监督机制与网络安全防护要求。制造商同时是AIA意义下的高风险AI系统提供者(Provider),需履行技术文件、日志记录、上市后监测及重大变更管理等责任。
欧盟授权代表(AR)是境外企业配置的关键角色,负责在欧盟境内承担法律责任,与主管机关沟通,并确保技术文件、合格声明与CE标志符合要求。AIA虽未强制设立AR,但NB在审查中通常要求海外企业通过AR履行境内责任。
进口商与分销商负责检查产品是否标注CE标志、是否完成EUDAMED注册、标签与UDI是否符合MDR/IVDR要求,以及在供应链中监督产品安全性。任何对产品合规性的疑问都需上报主管机关并与制造商协作处理。
此外,根据MDR/IVDR第15条,制造商还需设立合规负责人(PRRC),监督合格评定、上市后监测和技术文件的持续维护。
上述角色构成中国企业进入欧盟市场的最低配置。
(三)其他战略建议
MDAI的欧盟合规不仅依赖技术达标,对企业提出了体系化治理能力的更高要求。
首先,AIA要求制造商建立覆盖模型开发、训练、验证、测试、数据治理、日志记录、透明度与偏倚控制的增强型质量管理体系(QMS),中国企业应尽早在ISO 13485的基础上整合AIA要求,形成统一的AI合规体系。
其次,鉴于NB在审查过程中常遇到数据代表性不足、透明度不充分、模型更新路径不清晰等问题,企业需提前准备技术文件草案,与公告机构建立早期沟通机制,以降低审查时间与不符合风险。同时,应构建MDR Annex II/III、AIA Annex IV、ISO 13485与相关IEC标准的技术文件合规矩阵,确保文档结构一致、逻辑完整、可追溯性明确,从根本上提高NB审查效率。
此外,AIA对数据治理的要求强调数据集需具有与欧盟目标人群的一致性,中国企业应通过欧盟本地合作、多中心数据集或明确定义模型适用边界来提升模型可信度。
最后,对于持续学习型AI,应通过PDCP控制模型更新范围,避免触发实质性修改从而重新进行CE认证的风险,并在上市后建立数据漂移监测、偏倚监测与回滚机制,形成完整的闭环风险管理体系。
总体来看,随着MDR/IVDR与AIA的全面落地,欧盟监管范围已从传统产品安全扩展至算法治理、数据治理与系统透明度。中国企业越早完成法规映射、体系整合与NB前置沟通,就越能在未来的欧盟MDAI市场竞争中占据主动地位。
结语
随着MDR/IVDR与AIA的全面落地,欧盟已将MDAI带入一个兼具产品安全与算法治理的新监管阶段。企业在进入欧盟市场时,必须同时满足MDR/IVDR对安全性、性能与临床证据的要求,以及AIA对数据治理、透明度、人类监督与网络安全的系统性义务。
对于中国企业而言,这既是门槛,也是机遇。挑战在于,欧盟监管体系要求企业在产品层面之外,必须具备成熟的数据治理架构、模型验证体系、风险管理体系和透明度机制,单纯“补文档”已难以满足CE与AIA双重审查,企业必须在ISO 13485之上整合数据治理、模型验证、偏倚控制和上市后监测等机制,构建覆盖开发、部署到更新阶段的全链条AI治理合规体系。与此同时,AIA第8条赋予了一体化构建的空间,使企业可以通过统一的质量管理体系实现更高的合规效率。
面向未来,MDAI的竞争优势将不仅来自技术性能,更来自企业在风险控制、透明度、可追溯性及生命周期治理方面的能力。越早完成 MDR/IVDR与AIA的体系化整合,越能在欧盟市场中获得确定性、安全性与竞争性。合规不再只是准入门槛,而正在成为全球医疗AI产业的底层竞争力与长期成功的关键条件。
注 释:
1.《体外诊断医疗器械指令》(The In Vitro Diagnostic Medical Devices Directive, Directive 98/79/EC, “IVDD”)。
2.《医疗器械指令》(The Medical Devices Directive, Directive 93/42/EEC, “MDD”)。
3.MDR, Art. 2(42); IVDR, Art. 2(34).
4.一份由MedTech Europe发布的Market composition of in vitro diagnostic medical devices (IVDs)报告中指出,在旧IVDD下,仅约8%的IVD需要NB证书;而在IVDR下,需要NB认证的IVD占总量几乎80%。
https://www.medtecheurope.org/wp-content/uploads/2021/09/medtech-europe-survey-report-detailed-results.pdf
5.MDCG 2019-11 Rev.1: Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR, https://health.ec.europa.eu/document/download/b45335c5-1679-4c71-a91c-fc7a4d37f12b_en
6.AIB 20251 / MDCG 20256 Guidance: Interplay between MDR/IVDR and the AI Act, “The MDR and IVDR requirements address risks related to medical device software, however, they do not explicitly address risks specific to AI systems.” https://health.ec.europa.eu/document/download/b78a17d7-e3cd-4943-851d-e02a2f22bbb4_en?filename=mdcg_2025-6_en.pdf
7.新立法框架(New Legislative Framework, “NLF”)最早由Decision 768/2008/EC和Regulation (EC) 765/2008奠定立法基础,是欧盟产品(包括医疗器械)法规体系的基础性框架,它通过统一合格评估、标准化机制和市场监督等机制,使得不同法规能协调应用。https://single-market-economy.ec.europa.eu/single-market/goods/new-legislative-framework_en
8.AIA的附件一规定了“欧盟统一立法清单”,其中A节第11段和12段明确了包含MDR和IVDR。
9.MDR附录XVI产品主要包括MDR拓展覆盖的非医疗用途产品(如美学植入),一般需要NB介入。
10.MDR和IVDR第5条第5款均规定,在特定条件下,医疗机构可自制器械用于院内病患,仅用于该机构内部的诊断或治疗,不进行商业销售或转让,则无需取得CE认证或NB参与评定。
11.ISO 13485:2016 – Medical devices — Quality management systems — Requirements for regulatory purposes,国际标准化组织(ISO)发布的医疗器械领域专用质量管理体系标准。
12.AIA, Art. 13: “1. High-risk AI systems shall be designed and developed in such a way as to ensure that their operation is sufficiently transparent to enable deployers to interpret a system’s output and use it appropriately.”
13.AIA, Art. 50: “1. Providers shall ensure that AI systems intended to interact directly with natural persons are designed and developed in such a way that the natural persons concerned are informed that they are interacting with an AI system, unless this is obvious from the point of view of a natural person who is reasonably well-informed, observant and circumspect, taking into account the circumstances and the context of use.”
14.AIA, Art. 10: “3. Training, validation and testing data sets shall be relevant, sufficiently representative, and to the best extent possible, free of errors and complete in view of the intended purpose.”
15.AIA, Art. 12:
“1. High-risk AI systems shall technically allow for the automatic recording of events (logs) over the lifetime of the system.
2. In order to ensure a level of traceability of the functioning of a high-risk AI system that is appropriate to the intended purpose of the system, logging capabilities shall enable the recording of events relevant for: (a) identifying situations that may result in the high-risk AI system presenting a risk within the meaning of Article 79(1) or in a substantial modification; (b) facilitating the post-market monitoring referred to in Article 72; and (c) monitoring the operation of high-risk AI systems referred to in Article 26(5).”
16.MDR/IVDR Annex I.
17.AIA, Art. 14: “1. High-risk AI systems shall be designed and developed in such a way, including with appropriate human-machine interface tools, that they can be effectively overseen by natural persons during the period in which they are in use.”
18.AIA, Art. 14: “2. Human oversight shall aim to prevent or minimise the risks to health, safety or fundamental rights that may emerge when a high-risk AI system is used in accordance with its intended purpose or under conditions of reasonably foreseeable misuse, in particular where such risks persist despite the application of other requirements set out in this Section.”
19.AIA, Recitals (73): “...To that end, appropriate human oversight measures should be identified by the provider of the system before its placing on the market or putting into service. In particular, where appropriate, such measures should guarantee that the system is subject to in-built operational constraints that cannot be overridden by the system itself and is responsive to the human operator, and that the natural persons to whom human oversight has been assigned have the necessary competence, training and authority to carry out that role.”
20.MDCG 2019-16, Guidance on Cybersecurity for medical devices, https://mdi-europa.com/wp-content/uploads/2020/01/mdcg_2019_16_cybersecurity_en.pdf
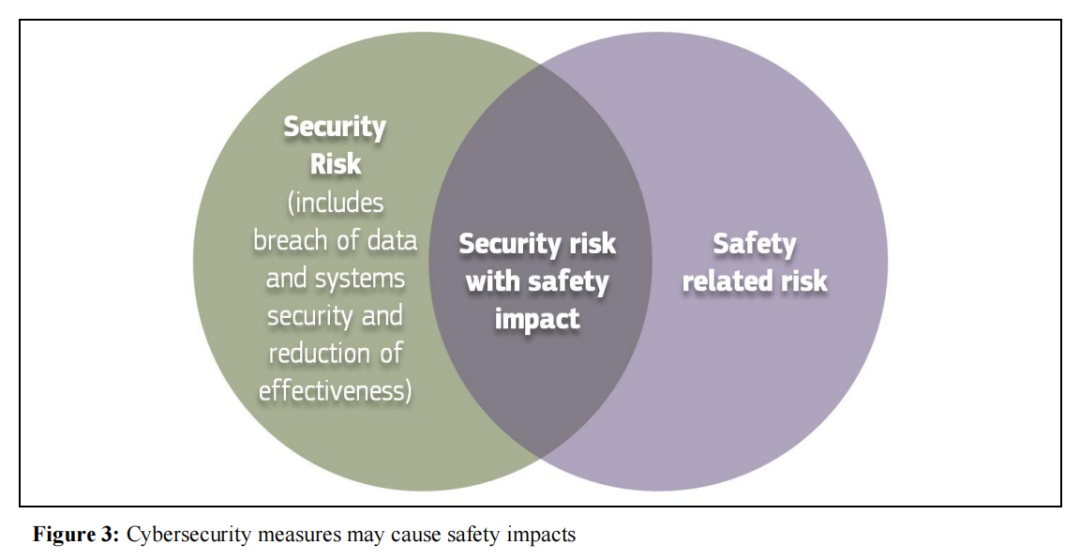
21.MDCG 2019-16的Figure 3表明,网络安全防护(security)风险与传统意义上的器械安全(safety)风险并非相互独立,而是存在显著的交叉与相互影响。当网络安全防护的脆弱性(如防护不足)或过度性(如防护过强)削弱了医疗器械的有效性、可用性或操控性时,此类网络安全防护风险将直接转化为具有器械安全影响的风险(security risk with safety impact)。基于MDR/IVDR附件I关于风险最小化与高水平健康安全保护的要求,制造商在进行风险管理时必须同时评估器械安全与网络安全防护之间的相互作用。
22.AIA Art. 15.
23.MDR Annex I Sec. 17, IVDR Annex I Sec. 16, MDCG 2019-16, AIA Recitals 76-77.
24.MDR Annex I Sec. 17, AIA Art. 15, AIA Annex IV Sec. 2.h.
25.AIA Art. 43.
(原标题:卓纬研究 | AI医疗器械全球合规系列(中篇)—— 欧盟篇:从MDR/IVDR到AIA的协同治理)
来源:北京卓纬律师事务所
作者:
- 李诘,北京卓纬律师事务所合伙人;业务领域:跨境投资与并购、政府事务与合规、数据安全与隐私保护、知识产权;联系电话:8621 6859 0516,电子邮箱:joseph.j.li@chancebridge.com
- 方云竹,北京卓纬律师事务所律师助理;业务领域:公司治理与跨境投资、政府事务与合规、网络安全与数据保护;联系电话:8621 6859 0516,电子邮箱:yunzhu.fang@chancebridge.com
特别声明:本微信公众号的文章仅供交流之用,不代表北京卓纬律师事务所或其律师的正式法律意见或建议。